临床前研究数据的合规性直接关系到药物、医疗器械等产品的上市审批,而与国际接轨是产品走向全球市场的关键。杭州环特生物科技股份有限公司严格遵循GLP(药物非临床研究质量管理规范)、OECD(经济合作与发展组织)等国际国内相关指导原则,确保临床前研究数据的真实性、完整性与合规性。在临床前研究过程中,建立完善的质量保证体系,对实验设计、操作流程、数据记录等环节进行全程管控;同时,采用国际认可的实验方法与检测标准,确保研究数据在全球范围内的认可度。此外,环特生物还为企业提供临床前研究数据的国际化申报咨询服务,帮助企业解决不同国家和地区审批要求的差异问题。其合规化、国际化的临床前研究服务,为企业产品的国内外上市提供了有力保障。临床前药效学研究可准确评估药物的医疗潜力。云南生物医药临床前安全性
营养保健食品的“蓝帽”备案注册离不开规范的临床前研究,其关键价值在于通过科学实验验证产品的功效与安全性。杭州环特生物科技股份有限公司构建了完善的临床前研究体系,为保健食品企业提供涵盖24项允许声称功能的检测服务。在临床前功效验证中,通过斑马鱼模型、哺乳动物模型等多种实验体系,量化评估产品的抗氧化、辅助降血脂、增强人体免疫能力等功效,确保功效宣称有充分的科学依据;安全性评价则聚焦急性经口毒性、遗传毒性等指标,排查产品潜在风险,保障消费者食用安全。临床前研究的数据不仅是产品备案的硬性要求,更是企业赢得市场信任的核心竞争力。环特生物凭借专业的临床前研究能力,帮助企业高效完成备案流程,推动产品快速合规上市。深圳化学药临床前一般毒理性评价临床前实验是成果转化桥梁,环特生物打通研发关键链路。

生物大分子药物(如抗体、蛋白、核酸等)因其高特异性和强的效性,已成为现代医药研发的关键方向。然而,其临床前研究面临独特挑战:分子量大导致膜通透性差、免疫原性风险高、稳定性控制难,且需针对特定靶点设计复杂作用机制。例如,单克隆抗体需通过抗体依赖细胞介导的细胞毒性(ADCC)或补体依赖细胞毒性(CDC)发挥作用,而双特异性抗体则需同时结合两个抗原表位以实现精细调控。临床前阶段需系统评估这些分子的药代动力学(PK)、药效动力学(PD)及毒性特征,通常采用体外细胞模型(如HEK293、CHO细胞)和体内动物模型(如小鼠、非人灵长类)相结合的策略。数据显示,全球生物大分子药物临床前研发失败率高达40%,其中因免疫原性或药代动力学问题导致的淘汰占比超60%,凸显了临床前研究的重要性。
临床前研究的起点是体外活性筛选,通过高通量技术(如96孔板、自动化液体处理系统)从化合物库中筛选出对靶点具有抑制或活动作用的“苗头化合物”。例如,针对EGFR突变型肺ancer,通过酶联免疫吸附试验(ELISA)筛选能抑制EGFR激酶活性的小分子,初始命中率可能低至0.1%。随后,通过构效关系(SAR)研究优化分子结构——通过合成系列类似物(如改变苯环取代基、调整酰胺键位置),结合表面等离子共振(SPR)技术测定结合亲和力(KD值),逐步提升活性(如将IC50从μM级优化至nM级)。这一阶段需平衡活性与理化性质(如logP、溶解度),避免“活性陷阱”(如过度追求高亲和力导致代谢不稳定)。例如,某候选HER2抑制剂通过引入氟原子降低脂溶性,成功将半衰期从2小时延长至8小时,为后续体内研究奠定基础。完善的质量体系,保障临床前实验全过程符合行业规范。
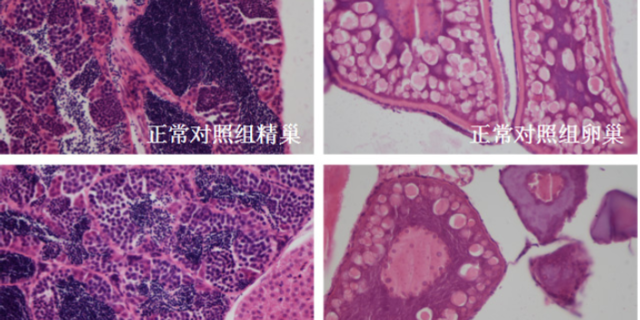
体外活性需通过体内模型验证其医疗潜力。根据疾病类型选择合适的动物模型是关键:例如,针对自身免疫病,常用NOD小鼠或胶原诱导性关节炎(CIA)模型;针对tumor,则采用患者来源异种移植(PDX)模型或基因工程小鼠(如KRAS突变型肺ancer模型)。以抗纤维化药物为例,将候选分子(如TGF-β1抑制剂)通过腹腔注射给予博来霉素诱导的肺纤维化小鼠,通过Micro-CT扫描量化肺密度变化,结合羟脯氨酸含量测定评估胶原沉积,可明确药物能否逆转纤维化进程。体内实验需设置严格对照组(如阳性的药、溶剂对照),并采用盲法评估以减少偏差。若候选分子在动物模型中显示出剂量依赖性疗效(如降低tumor体积30%以上),且效果优于或非劣于已上市药物,则可推进至毒理学研究。准确的临床前药效分析,助力企业筛选出潜力候选药物。药物临床前研究时间
临床前模型构建技术,是环特生物的主要竞争优势之一。云南生物医药临床前安全性
新药临床前毒理学研究是药物开发中保障患者安全的关键环节,其目标是通过系统评估候选药物对实验动物的毒性效应,预测其可能对人体产生的危害,为临床试验的剂量选择、风险控制及后续开发决策提供科学依据。这一阶段的研究需覆盖急性毒性(单次高剂量暴露)、重复给药毒性(多剂量、长期暴露)、遗传毒性(致突变性)、生殖毒性(致畸性、胚胎毒性)及特殊毒性(如光毒性、心脏毒性)等多个维度。据统计,全球约40%的新药在临床前毒理学阶段因安全性问题被淘汰,凸显其“安全阀”作用。例如,某抗tumor候选药物因在犬重复给药毒性实验中发现严重肝坏死,被迫终止开发,避免了潜在的临床肝衰竭风险。毒理学数据的可靠性直接决定了药物能否进入临床试验,其研究设计需严格遵循GLP(良好实验室规范)标准,确保数据的可重复性和监管认可。云南生物医药临床前安全性