急性毒性研究通过单次高剂量给药(如口服、静脉注射),测定药物的半数致死量(LD50)或比较大耐受剂量(MTD),明确其急性毒性阈值。例如,某中枢的神经系统药物在大鼠急性毒性实验中,LD50为500mg/kg,而MTD为200mg/kg,提示临床试验起始剂量应低于100mg/kg(通常为MTD的1/2-1/3)。重复给药毒性研究则通过多剂量、长期(如28天、90天)给药,观察靶organ毒性(如肝、肾、心脏)及剂量-毒性关系。以抗纤维化药物为例,在90天重复给药毒性实验中,犬在300mg/kg/天剂量下出现肾小管坏死,而100mg/kg/天剂量下无明显异常,提示临床安全剂量应≤100mg/kg。此类研究需结合病理学(HE染色、免疫组化)和临床病理学(血常规、生化指标)分析,明确毒性靶organ及可逆性(如停药后是否恢复)。临床前阶段的严格把关能提升新药上市的成功率。浙江天然药物临床前安全性

环特生物通过转化医学研究将临床前数据与临床需求紧密衔接,例如基于斑马鱼模型筛选的抗纤维化候选分子,在临床前研究中显示出对肺、肝纤维化的明显改善作用,其作用机制(抑制TGF-β1/Smad通路)与临床生物标志物(羟脯氨酸含量)高度相关,为后续临床试验设计提供了科学依据。在监管科学领域,环特参与制定了多项斑马鱼实验技术标准,其开发的“斑马鱼模型在药物心脏安全性评价中的应用”团体标准已被NMPA纳入创新药申报指南。此外,环特与FDA、EMA等监管机构保持密切沟通,通过提供符合GLP规范的斑马鱼及类organ数据,支持“条件性批准”或“快速通道”申请,例如某抗tumor双抗药物凭借环特提供的斑马鱼药效及安全性数据,获得FDA突破性疗法认定,研发周期缩短18个月。未来,环特将持续深化“临床前-临床”数据整合平台建设,推动创新药开发从“经验驱动”向“数据驱动”转型。湖北nmpa临床前评价机构环特生物累计完成八千 + 项目,临床前实验经验丰富。

药代动力学(PK)研究通过测定血药浓度-时间曲线,明确分子的吸收(如口服生物利用度)、分布(如血脑屏障穿透性)、代谢(如CYP450酶代谢途径)及排泄(如肾清理率)特性。例如,针对中枢的神经系统疾病候选分子,需通过脑脊液采样评估其能否穿透血脑屏障(脑/血比值>0.1为达标);针对口服药物,则需优化剂型(如纳米晶、自微乳化系统)以提升生物利用度(从<10%提升至>50%)。转化医学研究则进一步将临床前数据与临床关联——例如,通过肝微粒体孵育实验发现候选分子主要经CYP3A4代谢,提示其可能与利福平(CYP3A4诱导剂)存在药物相互作用风险,需在临床试验中设置药物相互作用研究组。此外,基于生理的药代动力学模型(PBPK)可预测不同人群(如儿童、肝肾功能不全患者)的PK特征,为临床剂量设计提供依据。这一阶段的成功标志是获得“安全、有效、可预测”的PK/PD(药效动力学)特征,为Ⅰ期临床试验的剂量爬坡设计提供关键参数。
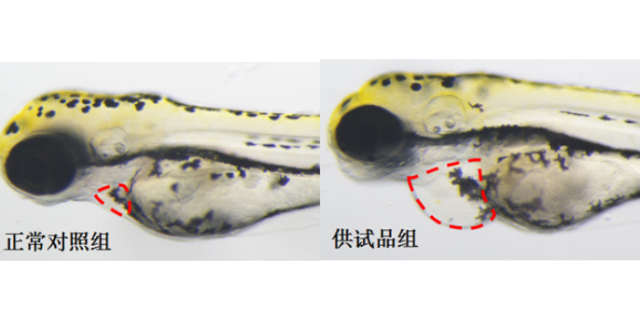
药效评估是判断化合物是否具备临床医疗价值的关键步骤。在临床前阶段,会构建多种疾病动物模型,如tumor移植模型、炎症模型、心血管疾病模型等,模拟人类疾病状态。以抗糖尿病化合物为例,通过给糖尿病模型小鼠灌胃或注射该化合物,监测其血糖水平、胰岛素分泌量、糖化血红蛋白等指标的变化。同时设置阳性对照组(使用已上市的同类药物)和阴性对照组(给予安慰剂)进行对比。若实验结果显示化合物能有效降低模型动物的血糖水平,且效果与阳性的药物相当或更优,同时不产生严重不良反应,那么该化合物就展现出良好的药效潜力。通过多方面、严谨的药效评估,筛选出真正具有医疗效果的化合物,推动其进入临床研究阶段,为后续临床试验的顺利开展提供有力支撑。临床前安全性评价是新药研发流程中不可或缺的关键环节。
药代动力学(PK)研究聚焦药物在体内的吸收、分布、代谢和排泄(ADME)过程,是决定药物剂量的关键。体外实验中,Caco-2细胞模型可预测药物肠道渗透性,肝微粒体或肝细胞孵育系统则用于评估代谢稳定性。例如,某候选抗ancer药物在肝微粒体中半衰期15分钟,提示需结构优化以提高代谢稳定性。活的体PK研究依赖大鼠或犬模型,通过液相色谱-质谱联用技术(LC-MS)测定血浆、组织中的药物浓度。环特生物开发的斑马鱼PK模型,可实时观察药物在胚胎体内的分布,发现某化合物在脑部的蓄积量是血浆的3倍,提示其可能穿透血脑屏障。PK/PD(药效动力学)整合分析进一步关联药物浓度与疗效,例如在antibiotic研发中,通过PK模型确定给药间隔,使血药浓度维持在小抑菌浓度(MIC)以上,显著提高杀菌效果。环特生物的临床前服务满足生物医药企业的多样需求。杭州毒理实验临床前新药评价中心项目
临床前研究可有效降低新药研发风险,提升研发成功率。浙江天然药物临床前安全性
遗传毒性研究通过Ames试验(细菌回复突变)、小鼠淋巴瘤试验(TK基因突变)及染色体畸变试验(如中国仓鼠卵巢细胞试验),评估药物是否可能引发基因突变或染色体损伤,从而增加ancer或遗传病风险。例如,某化疗药物在Ames试验中呈阳性,提示其可能具有致ancer性,需在临床试验中设置长期随访监测。生殖毒性研究则覆盖胚胎-胎仔发育毒性(EFD)、围产期毒性及两代的生殖毒性,评估药物对生育力、胚胎发育及后代的影响。以抗癫痫药物为例,在EFD实验中,大鼠在50mg/kg/天剂量下出现胎仔脊柱裂,提示育龄女性用药需严格避孕。此类研究需遵循ICH(国际人用药品注册技术协调会)S5指南,确保数据满足全球监管要求(如FDA、EMA)。浙江天然药物临床前安全性