临床前研究数据的合规性直接关系到药物、医疗器械等产品的上市审批,而与国际接轨是产品走向全球市场的关键。杭州环特生物科技股份有限公司严格遵循GLP(药物非临床研究质量管理规范)、OECD(经济合作与发展组织)等国际国内相关指导原则,确保临床前研究数据的真实性、完整性与合规性。在临床前研究过程中,建立完善的质量保证体系,对实验设计、操作流程、数据记录等环节进行全程管控;同时,采用国际认可的实验方法与检测标准,确保研究数据在全球范围内的认可度。此外,环特生物还为企业提供临床前研究数据的国际化申报咨询服务,帮助企业解决不同国家和地区审批要求的差异问题。其合规化、国际化的临床前研究服务,为企业产品的国内外上市提供了有力保障。临床前安全性评价是新药研发流程中不可或缺的关键环节。杭州临床前模式动物

药效评估是判断化合物是否具备临床医疗价值的关键步骤。在临床前阶段,会构建多种疾病动物模型,如tumor移植模型、炎症模型、心血管疾病模型等,模拟人类疾病状态。以抗糖尿病化合物为例,通过给糖尿病模型小鼠灌胃或注射该化合物,监测其血糖水平、胰岛素分泌量、糖化血红蛋白等指标的变化。同时设置阳性对照组(使用已上市的同类药物)和阴性对照组(给予安慰剂)进行对比。若实验结果显示化合物能有效降低模型动物的血糖水平,且效果与阳性的药物相当或更优,同时不产生严重不良反应,那么该化合物就展现出良好的药效潜力。通过多方面、严谨的药效评估,筛选出真正具有医疗效果的化合物,推动其进入临床研究阶段,为后续临床试验的顺利开展提供有力支撑。宁波药品临床前评价机构环特生物配备专业实验室,保障临床前研究高效开展。
对新药临床前毒理学研究结果的准确分析与解读直接关系到新药研发的决策。在分析毒理学数据时,首先要关注各项观察指标的变化趋势,不仅只是某个时间点的数值。例如在长期毒性试验中,体重变化曲线若呈现持续下降趋势,可能意味着药物对动物的营养代谢产生了不良影响,需要进一步深入分析原因。同时,要对不同剂量组之间的数据进行对比,判断剂量 - 反应关系是否存在。若随着药物剂量的增加,毒性反应的发生率和严重程度也相应增加,那么这种剂量 - 反应关系的存在提示药物毒性与剂量密切相关。此外,对于一些异常的检测结果,不能孤立地看待,需要综合考虑动物的整体状况、其他相关指标的变化以及试验过程中的各种因素。例如,血液生化指标中某个酶活性的升高,可能是药物直接作用于肝脏细胞导致,也可能是由于动物在试验过程中受到应激等其他因素干扰。只有通过多方面、系统、科学的分析与解读,才能从毒理学研究结果中提炼出真实、准确的信息,为新药能否进入临床试验以及后续的研发方向提供合理的判断依据。
罕见病药物研发因病例稀少、研究基础薄弱,其临床前研究面临诸多挑战,而高效的临床前研究体系是突破这些瓶颈的关键。杭州环特生物科技股份有限公司针对罕见病的特点,构建了专属的临床前研究平台,为罕见病药物研发提供技术支撑。在临床前模型构建方面,通过基因编辑技术构建罕见病特异性斑马鱼模型与哺乳动物模型,模拟疾病的病理特征,解决罕见病模型匮乏的问题;在药物筛选中,利用斑马鱼模型的高通量优势,快速筛选潜在医疗药物,缩短研发周期。此外,临床前研究还需加强安全性评价的深度,避免因罕见病患者群体的特殊性导致的潜在风险。环特生物的临床前研究服务,为罕见病药物研发降低了门槛、提高了效率,为罕见病患者带来新的医疗希望。环特生物以临床前研究为关键,赋能医药创新发展。

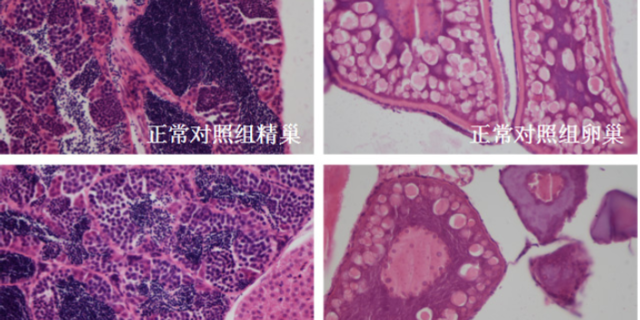
急性毒性研究通过单次高剂量给药(如口服、静脉注射),测定药物的半数致死量(LD50)或比较大耐受剂量(MTD),明确其急性毒性阈值。例如,某中枢的神经系统药物在大鼠急性毒性实验中,LD50为500mg/kg,而MTD为200mg/kg,提示临床试验起始剂量应低于100mg/kg(通常为MTD的1/2-1/3)。重复给药毒性研究则通过多剂量、长期(如28天、90天)给药,观察靶organ毒性(如肝、肾、心脏)及剂量-毒性关系。以抗纤维化药物为例,在90天重复给药毒性实验中,犬在300mg/kg/天剂量下出现肾小管坏死,而100mg/kg/天剂量下无明显异常,提示临床安全剂量应≤100mg/kg。此类研究需结合病理学(HE染色、免疫组化)和临床病理学(血常规、生化指标)分析,明确毒性靶organ及可逆性(如停药后是否恢复)。完善的质量体系,保障临床前实验全过程符合行业规范。宁波药品临床前评价机构
环特生物深耕临床前实验,为药物研发筑牢早期研究基础.杭州临床前模式动物
毒理学研究是临床前研究的“安全阀”,需通过急性毒性(单次高剂量给药)、重复给药毒性(28天/90天多次给药)、遗传毒性(Ames试验、小鼠淋巴瘤试验)及生殖毒性(胚胎致死性、致畸性)实验,多方面评估分子的安全性。例如,针对抗凝血药物,需通过大鼠尾静脉出血时间实验确定耐受剂量(MTD),避免引发过度出血风险;针对抗tumor药物,则需关注骨髓抑制(通过血常规检测白细胞、血小板计数)及肝毒性(通过ALT/AST酶活性测定)。特殊毒性研究(如光毒性、心脏毒性)也不可忽视——例如,通过hERG通道抑制实验评估药物是否可能引发QT间期延长。毒理学数据的解读需结合医疗窗(有效剂量与毒性剂量的比值),若候选分子的医疗指数(TI)>5,则认为安全性可控;若TI<3,则需重新优化结构或调整给药的方案。杭州临床前模式动物