- 品牌
- 司鼎;OriCell
Real-time PCR原理:基于探针的化学法,Real-time PCR应用一个或多个荧光标记的寡核苷酸探针检测PCR扩增产物;依赖荧光能量共振传递(FRET)检测特异性扩增产物,单单检测特异性扩增产物,DNA及RNA定量;基因表达验证;等位基因鉴别;SNP分型;病原体和病毒检测;多重PCR,探针和目标片段的特异性结合产生荧光信号,因此减少了背景荧光和假阳性;探针可标记不同波长的荧光基团,用于多重PCR反应。对于不同的靶序列需要合成不同的探针,原料成本较高。PCR扩增时在加入一对引物的同时加入一个特异性的萤光探针。厦门微量荧光定量PCR设计公司

Real-time PCR链反应的常见问题分析与解决方法:反应缓冲液未完全融化或未充分混匀。确保反应缓冲液融化完全并彻底混匀。引物特异性差。利用BLAST检查引物特异性或重新设计引物。引物量过多。减少反应体系中引物的用量。模板量过多。质粒DNA的用量应<50ng,而基因组DNA则应<200ng。外源DNA污染。确保操作的洁净。阴性对照出现条带:试剂,头,工作台污染。使用全新的试剂和头,对工作台进行清洁。条带大小与理论不符:污染。使用全新的试剂和头,对工作台进行清洁。模板或引物使用错误。更换引物和模板。基因亚型。对研究的基因进行序列分析和BLAST研究。逆转录-聚合酶链反应较广用于表达谱,以确定基因的表达或鉴定RNA转录物的序列。莆田骨头数字PCR研究方案聚合酶链式反应又称多聚酶链式反应。
Real-time PCR实验注意事项-防止RNA酶污染的措施:1.所有的玻璃器皿均应在使用前于180℃的高温下干烤6hr或更长时间。2.塑料器皿可用0.1%DEPC水浸泡或用氯仿冲洗(注意:有机玻璃器具因可被氯仿腐蚀,故不能使用)。3.有机玻璃的电泳槽等,可先用去污剂洗涤,双蒸水冲洗,乙醇干燥,再浸泡在3%H2O2室温10min,然后用0.1%DEPC水冲洗,晾干。4.配制的溶液应尽可能的用0.1%DEPC,在37℃处理12hr以上。然后用高压灭菌除去残留的DEPC。不能高压灭菌的试剂,应当用DEPC处理过的无菌双蒸水配制,然后经0.22μm滤膜过滤除菌。5.操作人员戴一次性口罩、帽子、手套,实验过程中手套要勤换。6.设置RNA操作专业使用实验室,所有器械等应为专业使用。
Real-time PCR萤光染料掺入法:在PCR反应体系中,加入过量SYBR萤光染料,SYBR萤光染料特异性地掺入DNA双链后,发射萤光信号,而不掺入链中的SYBR染料分子不会发射任何萤光信号,从而保证萤光信号的增加与PCR产物的增加完全同步。此方法适用于:1、灵敏度高:使用SYBR可使萤光效果增强到1000倍以上。2、通用性好,不需要设计探针,方法简便,省时,价格低廉。3、通用型方法,在国内外科研中普遍使用。4、高通量大规模的定量PCR检测。5、专一性要求不高的定量PCR检测。Real-time PCR提高实验的重复性。

Real-time PCR链式反应的常见问题:靶序列变异:如靶序列发生突变或缺失,影响引物与模板特异性结合,或因靶序列某段缺失使引物与模板失去互补序列,其PCR扩增是不会成功的。假阳性:出现的PCR扩增条带与目的靶序列条带一致,有时其条带更整齐,亮度更高。引物设计不合适:选择的扩增序列与非目的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非目的性的序列。靶序列太短或引物太短,容易出现假阳性。需重新设计引物。出现片状拖带或涂抹带:PCR扩增有时出现涂抹带或片状带或地毯样带。其原因往往由于酶量过多或酶的质量差,dNTP浓度过高,Mg2+浓度过高,退火温度过低,循环次数过多引起。其对策有:减少酶量,或调换另一来源的酶。②减少dNTP的浓度。适当降低Mg2+浓度。增加模板量,减少循环次数。聚合酶链式反应准备:PCR所用的酶主要有两种来源:Taq和Pfu,分别来自两种不同的噬热菌。定量聚合酶链反应或者实时聚合酶链反应不要与逆转录允许估计样品中存在的给定序列的量。厦门微量荧光定量PCR设计公司
Real-time PCR反应的一个主要限制是,为了产生其选择性扩增的引物,需要目标序列的先前信息。厦门微量荧光定量PCR设计公司
Real-time PCR链式反应:每次循环都得加入新的DNA聚合酶,不但操作烦琐,而且价格昂贵,制约了PCR技术的应用和发展。耐热DNA聚合酶-Taq酶的发现对于PCR的应用有里程碑的意义,该酶可以耐受90℃以上的高温而不失活,不需要每个循环加酶,使PCR技术变得非常简捷、同时也降低了成本,PCR技术得以大量应用,并逐步应用于临床。多重连接依赖探针扩增允许用单个引物对扩增多个靶标,从而避免多重PCR的分辨率限制。引物的设计注意事项:1,注意引物设计好后的产物长度。Real-time PCR的较佳产物长度在150-250kb左右(书上说这样可以增加荧光的敏感度,减少实验误差,但是我对这个说法持怀疑态度。产物长度越大,SYBR嵌入的越多,这样才能够保证之后的荧光值比较可信)。2,不同的管家基因扩增效率不同。所以在做整个实验之前应该做一次检测扩增效率的实验。如果扩增效率相差太大,就不能使用delt,deltCt法来比较基因表达量的高低。厦门微量荧光定量PCR设计公司
上海司鼎生物科技有限公司成立于2016-06-07年,在此之前我们已在免疫印迹(WB)技术服务,荧光定量PCR技术服务,膜片钳电生理技术服务,在体光纤成像记录技术服务行业中有了多年的生产和服务经验,深受经销商和客户的好评。我们从一个名不见经传的小公司,慢慢的适应了市场的需求,得到了越来越多的客户认可。公司主要经营免疫印迹(WB)技术服务,荧光定量PCR技术服务,膜片钳电生理技术服务,在体光纤成像记录技术服务等产品,我们依托高素质的技术人员和销售队伍,本着诚信经营、理解客户需求为经营原则,公司通过良好的信誉和周到的售前、售后服务,赢得用户的信赖和支持。公司与行业上下游之间建立了长久亲密的合作关系,确保免疫印迹(WB)技术服务,荧光定量PCR技术服务,膜片钳电生理技术服务,在体光纤成像记录技术服务在技术上与行业内保持同步。产品质量按照行业标准进行研发生产,绝不因价格而放弃质量和声誉。在市场竞争日趋激烈的现在,我们承诺保证免疫印迹(WB)技术服务,荧光定量PCR技术服务,膜片钳电生理技术服务,在体光纤成像记录技术服务质量和服务,再创佳绩是我们一直的追求,我们真诚的为客户提供真诚的服务,欢迎各位新老客户来我公司参观指导。

引物的设计注意事项:1,引物设计要跨内含子,不能在一个外显子上(我们的实验是以cDNA为模板来扩增的,如果有DNA污染的话,因为DNA上含有分子量很大的内含子,不可能完成扩增。这对我们消除整个实验的误差起很大的作用)。2,引物设计的时候要尽可能设计在同一退火温度,方便于以后同时扩增很多不同的基因片段。但是如果相差比较大,就得分开做,浪费模板,浪费管家基因,所以每个实验室设计引物的时候要尽可能一致,这样就不用每次查退火温度,且对整个实验有利。在不同的PCR反应管中加入已定量的内标和引物,内标用基因工程方法合成。苏州微量RT-PCR检测技术原理及步骤聚合酶链式反应准备:PCR引物设计,PCR反应中...
- 上海骨头Real-time PCR 2024-12-26
- 武汉实时数字PCR哪家好 2024-12-25
- 南京实时荧光定量PCR 2024-12-25
- 苏州骨头定量PCR设计公司 2024-12-25
- 上海特殊样本数字PCR供应商 2024-12-25
- 连云港实时数字PCR服务 2024-12-25
- 南京特殊样本数字PCR原理 2024-12-25
- 温州特殊样本PCR检测技术设计公司 2024-12-25
- 无锡细胞定量PCR原理及步骤 2023-08-11
- 温州分子生物学Real-time PCR应用 2023-08-11


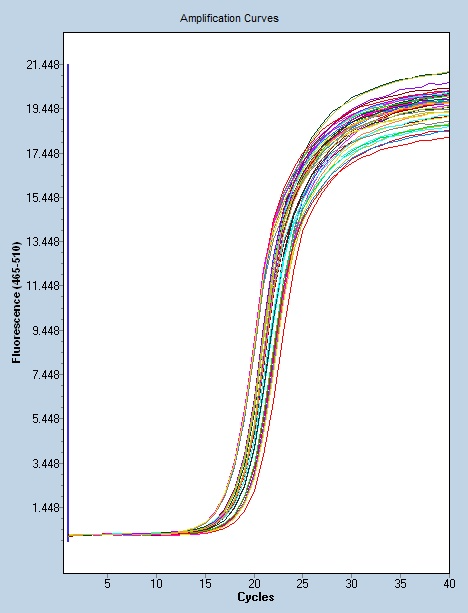
- 广州微量数字PCR供应商 2023-08-10
- 连云港血液RT-PCR检测技术服务 2023-08-09
- 温州细胞PCR检测技术服务 2023-08-09
- 徐州细胞RT-PCR检测技术供应商 2023-08-09
- 苏州微量数字PCR供应商 2023-08-08
- 珠海分子生物学荧光定量PCR网站 2023-08-08

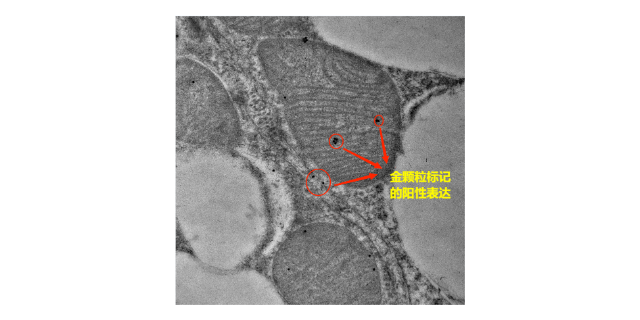
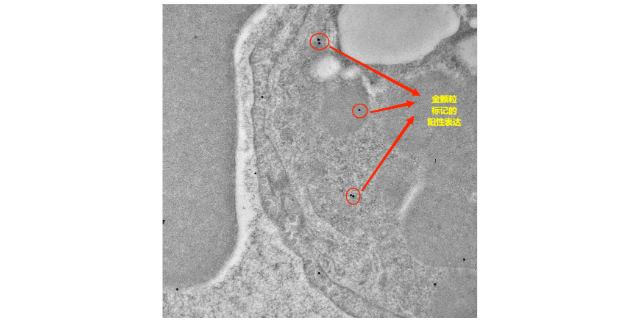
- 黄石细菌免疫电镜技术用途 01-05
- 绍兴亚细胞水平免疫电镜检测哪里有 01-05
- 襄阳抗体反应免疫电镜检测特点 01-05
- 绍兴病毒免疫电镜技术原理 01-05
- 芜湖细菌免疫电镜技术应用 01-05
- 深圳抗体反应免疫电镜技术哪里有 01-05
- 合肥免疫性疾病免疫电镜技术哪家靠谱 01-05
- 温州高灵敏度免疫电镜技术哪里有 01-05
- 南通抗原定位免疫电镜技术服务公司 01-05
- 淮南抗体反应免疫电镜检测 01-05