- 品牌
- 司鼎;OriCell
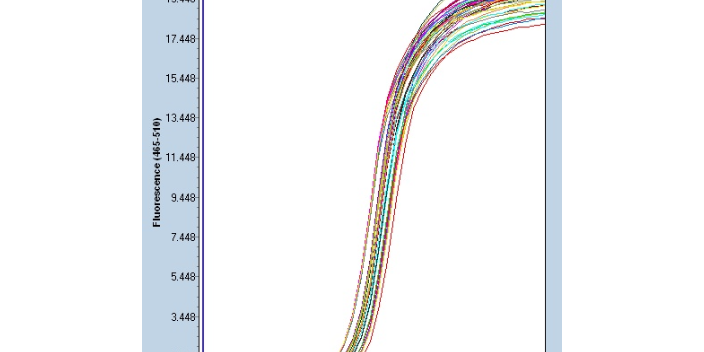
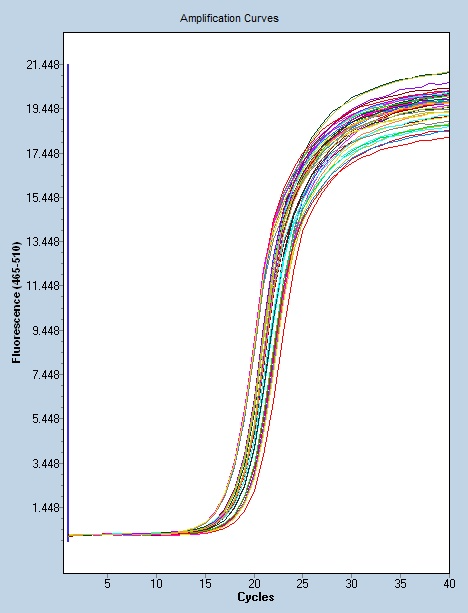
聚合酶链式反应的特点:灵敏度高:PCR产物的生成量是以指数方式增加的,能将皮克(pg=10-12)量级的起始待测模板扩增到微克(μg=-6)水平。能从100万个细胞中检出一个靶细胞;在病毒的检测中,PCR的灵敏度可达3个RFU(空斑形成单位);在细菌学中很小检出率为3个细菌。简便、快速:PCR反应用耐高温的Taq DNA聚合酶,一次性地将反应液加好后,即在DNA扩增液和水浴锅上进行变性-退火-延伸反应,一般在2~4 小时完成扩增反应。扩增产物一般用电泳分析,不一定要用同位素,无放射性污染、易推广。纯度要求低:不需要分离病毒或细菌及培养细胞,DNA 粗制品及RNA均可作为扩增模板。可直接用临床标本如血液、体腔液、洗嗽液、毛发、细胞、活组织等DNA扩增检测。聚合酶链反应可用于产生突变基因,突变由科学家随意选择。南京实时Real-time PCR方案

industryTemplate南京实时Real-time PCR方案聚合酶链反应可以用于分析病症、微生物或其他疾病状态中基因表达水平的变化。
聚合酶链反应的常见问题分析与解决方法:反应缓冲液未完全融化或未充分混匀。确保反应缓冲液融化完全并彻底混匀。引物特异性差。利用BLAST检查引物特异性或重新设计引物。引物量过多。减少反应体系中引物的用量。模板量过多。质粒DNA的用量应<50ng,而基因组DNA则应<200ng。外源DNA污染。确保操作的洁净。阴性对照出现条带:试剂,头,工作台污染。使用全新的试剂和头,对工作台进行清洁。条带大小与理论不符:污染。使用全新的试剂和头,对工作台进行清洁。模板或引物使用错误。更换引物和模板。基因亚型。对研究的基因进行序列分析和BLAST研究。
聚合酶链式反应的常见问题:Mg2+浓度:Mg2+离子浓度对PCR扩增效率影响很大,浓度过高可降低PCR扩增的特 异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。反应体积的改变:通常进行PCR扩增采用的体积为20ul、30ul、50ul。或100ul,应用多 大体积进行PCR扩增,是根据科研和临床检测不同目的而设定,在做小体积如20ul 后,再做大体积时,一定要模索条件,否则容易失败。物理原因:变性对PCR扩增来说相当重要,如变性温度低,变性时间短,极有可能出现假阴性;退火温度过低,可致非特异性扩增而降低特异性扩增效率退火温度过高影响引物与模板的结合而降低PCR扩增效率。有时还有必要用标准的温度计,检测一下扩增仪或水溶锅内的变性、退火和延伸温度,这也是PCR失败的原因之一。聚合酶链式反应中的DMSO、甘油等,主要用来保持酶的活性和帮助DNA解除缠绕结构。

聚合酶链式反应:Taq(水生栖热菌)聚合酶耐热DNA聚合酶的很好活性温度约为75-80°C(167-176°F),尽管该酶通常使用72°C(162°F)的温度。在该步骤中,DNA聚合酶通过从反应混合物中添加在5’-至-3’方向上与模板互补的游离DNA链来合成与DNA模板链互补的新DNA链,在新生(伸长)DNA链的末端,将dNTPs的5'-磷酸基与3'-羟基缩合。延伸所需的精确时间取决于所使用的DNA聚合酶和要扩增的DNA目标区域的长度。根据经验,在很好温度下,大多数DNA聚合酶每分钟聚合一千个碱基。在很好条件下(即,如果没有限制底物或试剂的限制),在每个延伸/延伸步骤中,DNA靶序列的数量加倍。随着每一个连续的循环,原始模板链加上所有新产生的链成为下一轮延伸的模板链,导致特定DNA目标区域的指数(几何)扩增。PCR由变性-退火-延伸三个基本反应步骤构成。南京实时Real-time PCR方案
许多疾病的未知病因的测序正在通过聚合酶链反应得到解决。南京实时Real-time PCR方案
聚合酶链式反应:反应的控制:PCR反应的缓冲液 提供合适的酸碱度与某些离子;镁离子浓度 总量应比dNTPs的浓度高,常用1.5mmol/L;底物浓度 dNTP以等摩尔浓度配制,20~200umol/L;TaqDNA聚合酶 2.5U(100ul);引物浓度一般为0.1 ~ 0.5umol/L;反应温度和循环次数;变性温度和时间 95℃,30s;退火温度和时间 低于引物Tm值5 ℃左右,一般在45~55℃;延伸温度和时间 72℃,1min/kb(10kb内);Tm值=4(G+C) +2(A+T);循环次数 :一般为25 ~ 30次。循环数决定PCR扩增的产量。模板初始浓度低,可增加循环数以便达到有效的 扩增量。但循环数并不是可以无限增加的。一般循环数为30个左右,循环数超过30个以后,DNA聚合酶活性逐渐达到饱和,产物的量不再随循环数的增加而增加,出现了所谓的“平台期”。南京实时Real-time PCR方案
引物的设计注意事项:1,引物设计要跨内含子,不能在一个外显子上(我们的实验是以cDNA为模板来扩增的,如果有DNA污染的话,因为DNA上含有分子量很大的内含子,不可能完成扩增。这对我们消除整个实验的误差起很大的作用)。2,引物设计的时候要尽可能设计在同一退火温度,方便于以后同时扩增很多不同的基因片段。但是如果相差比较大,就得分开做,浪费模板,浪费管家基因,所以每个实验室设计引物的时候要尽可能一致,这样就不用每次查退火温度,且对整个实验有利。在不同的PCR反应管中加入已定量的内标和引物,内标用基因工程方法合成。苏州微量RT-PCR检测技术原理及步骤聚合酶链式反应准备:PCR引物设计,PCR反应中...
- 上海骨头Real-time PCR 2024-12-26
- 武汉实时数字PCR哪家好 2024-12-25
- 南京实时荧光定量PCR 2024-12-25
- 苏州骨头定量PCR设计公司 2024-12-25
- 上海特殊样本数字PCR供应商 2024-12-25
- 连云港实时数字PCR服务 2024-12-25
- 南京特殊样本数字PCR原理 2024-12-25
- 温州特殊样本PCR检测技术设计公司 2024-12-25
- 无锡细胞定量PCR原理及步骤 2023-08-11
- 温州分子生物学Real-time PCR应用 2023-08-11


- 广州微量数字PCR供应商 2023-08-10
- 连云港血液RT-PCR检测技术服务 2023-08-09
- 温州细胞PCR检测技术服务 2023-08-09
- 徐州细胞RT-PCR检测技术供应商 2023-08-09
- 苏州微量数字PCR供应商 2023-08-08
- 珠海分子生物学荧光定量PCR网站 2023-08-08

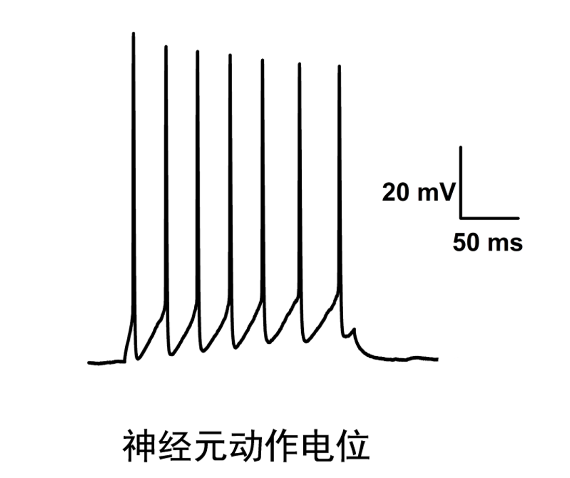

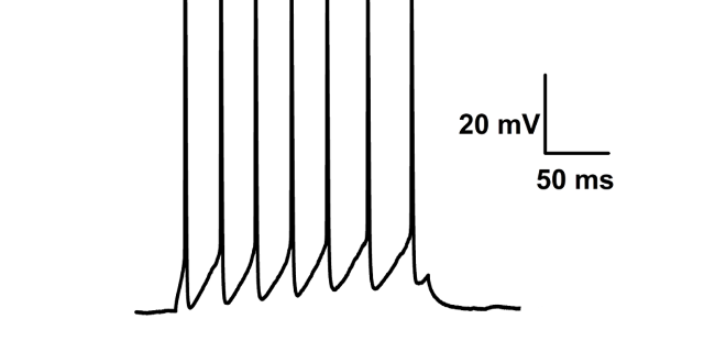
- 福州神经生物学膜片钳电生理技术应用 05-23
- 连云港神经生物学电生理膜片钳原理 05-22
- 莆田神经生物学膜片钳电生理技术方案 05-22
- 金华全自动膜片钳 05-22
- 湖州神经生物学脑片膜片钳服务 05-22
- 连云港全自动膜片钳技术 05-22
- 湖州细胞生物学实用膜片钳原理及步骤 05-22
- 东莞神经生物学膜片钳网站 05-22
- 徐州全自动膜片钳技术设计公司 05-22
- 苏州细胞生物学膜片钳网站 05-22